ASKE-E Month 5 Milestone Report¶
Semantic filters to improve model analysis¶
Examining the explanations produced by the COVID-19 EMMAA model for in-vitro drug screening experiments, we found that some of the explanations included causal mechanisms that were not consistent with the nature of the experimental context being studied. For instance, in an experiment where a single drug is added in a controlled manner, a mechanism that involves another drug (for instance via a drug-drug interaction) is not appropriate. Similarly, for an in-vitro experiment, higher-level societal factors are semantically not appropriate as intermediate concepts on a causal path.
Motivated by this, we implemented an approach to applying semantic filters to mechanistic paths that allow encoding constraints on what is and isn’t allowed on paths when explaining a given observation. These constraints derive from what is known about the experimental context in which that observation was made. For the observations used as test conditions for the COVID-19 model, we created constraints to exclude small molecules other than the drug that is used in the given experiment and higher level concepts including phenotypes, organisms and diseases. We found that the quality of explanations found improved substantially and is now more appropriate semantically with respect to the experimental context.
Model analysis exploiting ontological relationships¶
During this reporting period we extended the way EMMAA models are tested against experimental observations. Previously, we applied tests to models based on a strict match between the entities in the test and the set of entities in the model. However we noticed that in many cases models tended to consist of highly specific entities (e.g., individual proteins like KRAS, HRAS, and NRAS), whereas literature mining often picked up tests involving higher-level ontological concepts (e.g., the RAS protein family). The limitation of this approach was that we could only return a path based on exact matches between test and model entities, even when the model contained a path among more specific entities that would serve as a test explanation.
In the new approach we allow relations among more specific concepts to serve as explanations for relations among more general concepts (but not the reverse). Specificity is determined not only by hierarchical levels in the ontology (e.g. a member of a protein family is more specific than the family entity), but also by the amount of contextual information supplied for an entity (e.g., a protein with a phosphorylation is a more specific version of the same entity without a phosphorylation). This information is used to determine which tests can be applied to the model and also to find explanatory paths. To make this relationship explicit in our explanations, when a path found starts or ends with a more specific version of a test entity, we add a special “is a refinement of” or “has a refinement” edge to the path.
We applied this new testing approach to the EMMAA COVID-19 model. For the tests from the MITRE Therapeutic Information Browser Corpus (“MITRE Tests”), 174 new tests were determined to be relevant to the model when taking refinements into account. For these tests, which generally take the form “drug X inhibits virus Y”, we found relevant, more specific agents both for drugs (e.g., “rifampicin” is a type of “RNA polymerase inhibitor”) and viruses (e.g., “infectious bronchitis virus” is a type of “gammacoronavirus”). Of these new tests, 95 passed in the signed graph network.
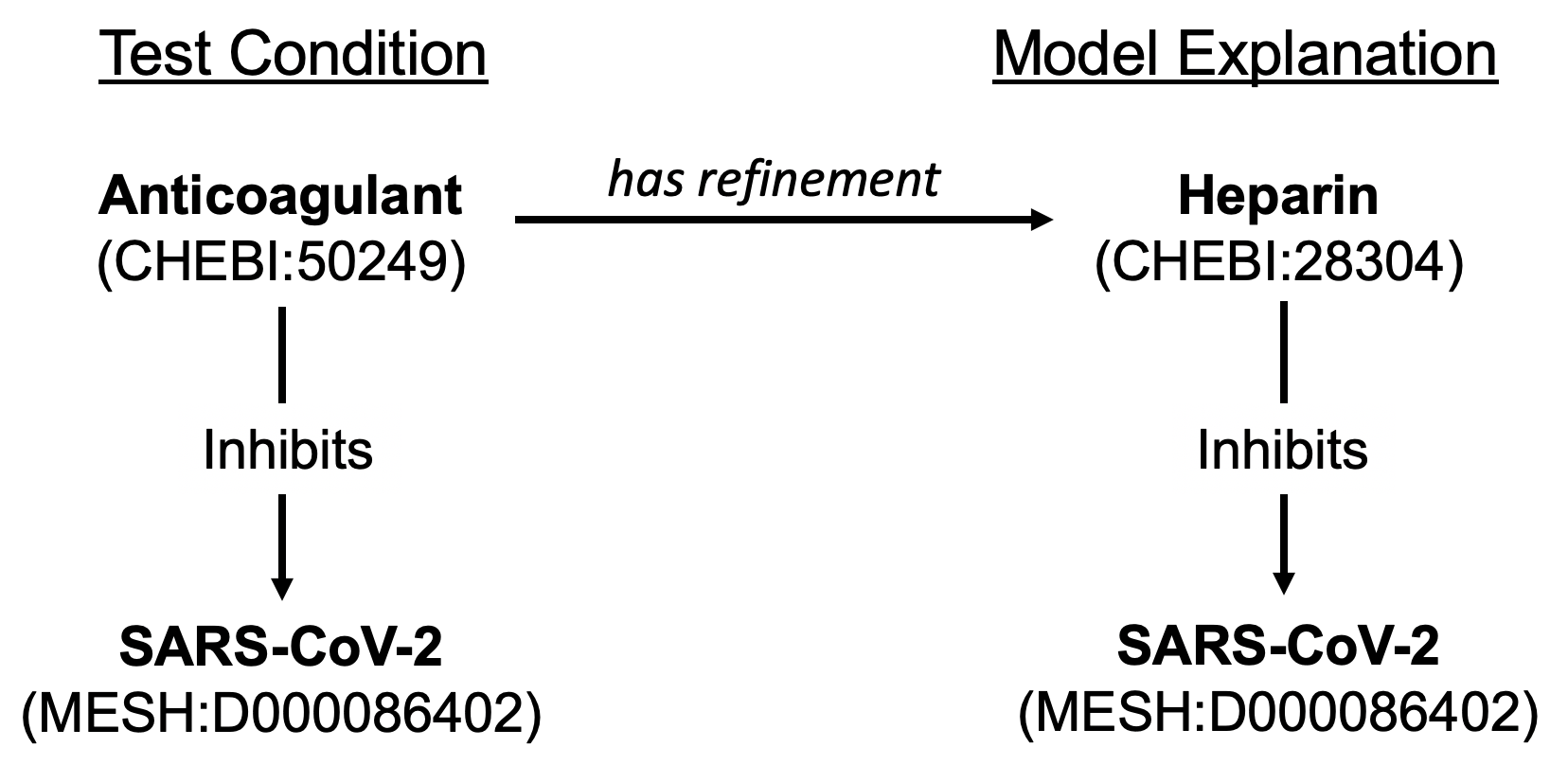
An example new passing test is shown in the figure below for the test condition “anticoagulant inhibits SARS-CoV-2”, which was previously determined to not be relevant to the model due to the fact that the model did not contain the specific entity for “anticoagulant” (CHEBI:50249). The model contains the information that heparin (CHEBI:28304), a type of anticoagulant, inhibits SARS-CoV-2, and the system now returns the explanation that “anticoagulant has refinement heparin; heparin inhibits SARS-CoV-2.”
Improved reading and assembly of protein chains and fragments¶
Protein chains and fragments are important both for human and viral biology. In ASKE-E month 2, we reported having extended the Reach reading system with lexicalizations of these entities from UniProt and the Protein Ontology (PR). This month, we made a number of extensions to our software stack to propagate these extensions in a useful way.
First, UniProt and PR have a large number of overlapping entries but neither source provides mappings to the other at the level of protein chains (only full protein entries). We developed a semi-automated approach to find and curate these mappings. We used Gilda to find lexical overlaps between the two ontologies and put these as predictions into the Biomappings repository and curation tool. We then curated these mappings to confirm correct ones and remove incorrect ones. These mappings were then propagated into the INDRA Ontology graph to be used for standardization.
Second, we found that the names of protein chains (similar to the names of full proteins) are ambiguous across organisms. This is especially problematic with the large number of viral species and strains that contain protein chains with identical or similar names. Current machine reading systems including Reach typically cannot disambiguate across these choices and produce highly ambiguous groundings for these viral proteins. Therefore, contextual information needs to be brought in externally to decide which organism to prioritize when selecting a grounding produced by Reach. To this end, we implemented an organism prioritization scheme whereby the user (or some external automated process) can supply a ranked list of organism identifiers to represent priority. This list is then used to guide how to the grounding of proteins and protein chains is selected. For example, if a paper is known to describe SARS-CoV-2 and human biology, one can supply an organism priority list including the identifiers of these two organisms to exclude or de-prioritize any spurious groundings from e.g., other viral strains that are irrelevant in the given context. Further, the organisms which a paper describes can be obtained from annotations that are either provided directly with the paper in PubMed or can be obtained using dedicated NLP systems set up for this task e.g, the MTI system.
Going forward, we will re-process the COVID-19 papers with these features in place and expect that the quality of reading, extraction and assembly for virus-host interactions will improve significantly.
Bio ontology optimized for visualization¶
We implemented a custom export of the INDRA BioOntology graph that is optimized for organizing nodes in a UI. The idea is to create top-level groups of entities that correspond to an intuitive category (e.g., human genes/proteins, non-human genes/proteins, small molecules, diseases, etc.). EMMAA models don’t contain this information about their entities directly, rather, they are inferred from identifiers assigned to each entity in a given set of name spaces. However, some name spaces contain multiple types of entities (e.g., MESH contains small molecules as well as diseases) and some entity types are distributed across multiple name spaces (e.g., human genes/proteins can be grounded to HGNC, UniProt, FamPlex, etc.). In this custom export, we split some name spaces and merged others to create a more ideal resolution and shared this export with the Uncharted team.