ASKE-E Month 12 Milestone Report¶
EMMAA and its role in the integrated architecture¶
By the end of this reporting period we completed our original integration plan for EMMAA with other systems developed in ASKE-E. We have reported on the technical details of each of these integrations in previous reports, here we summarize key points. The integration architecture is shown in the following diagram:
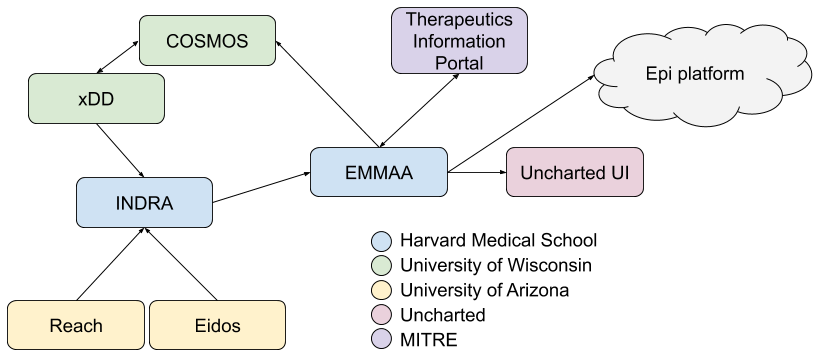
First, INDRA is integrated with (in addition to dozens of other knowledge sources) the Reach and Eidos machine reading systems developed by the University of Arizona team and we have made significant improvements to the utility of these systems tailored to ASKE use cases of interest such as modeling COVID-19 disease mechanisms.
We integrated INDRA and EMMAA with the xDD and COSMOS systems developed by the University of Wisconsin team. Machine reading systems integrated with INDRA are deployed to xDD and read new literature content each day such that this content complement the open-access content processed each day on our team’s compute infrastructure. Results are propagated back to INDRA and made use of by EMMAA for daily model updates. EMMAA is also integrated with COSMOS in several ways, first, it can access and integrated figures and tables extracted from a given publication through the COSMOS API; second, it can query for figures and tables relevant for a given pair of entities to provide figure/table context for statements in a given EMMAA model.
The EMMAA COVID-19 model is also integrated with the MITRE Therapeutics Information Portal, namely, EMMAA constructs model-based mechanistic explanations for observations on drug-coronavirus effects collected by TIP and makes these explanations browsable and auditable on the EMMAA dashboard.
EMMAA and all its models are also integrated with the Uncharted UI which draws on the data and APIs exposed by EMMAA, as well as the INDRA Ontology graph to render large networks, and drill down into smaller subnetworks in an integrated graphical interface.
EMMAA’s integration with the epidemiology platform centers around model representations: EMMAA supports executable model export into the GroMET format which is supported by the same simulation and analysis tools developed for epidemiology models. This integration at the executable model level is also compatible with the Uncharted UI, complementing EMMAA’s contributions at the larger-scale causal network level. Finally, another connection with epidemiological models is through the causal relations the COVID-19 EMMAA model collects, namely, EMMAA now also integrates causal relations between high-level concepts (diseases, phenotypes, etc.) in addition to molecular entities and cellular processes.
The current state of EMMAA¶
EMMAA currently makes 16 models available covering a wide range of scopes and conceptual modeling approaches. Some models are fully automatically assembled and center around a given disease area (e.g., Melanoma, Vitiligo, Neurofibromatosis) with their scope defined by a set of search terms (genes, etc.) into the scientific literature. Other models are pathway focused such as the Ras Model which cuts across multiple biological contexts and focuses on pathway mechanisms, its scope defined by members of the pathway. Several models are built using expert input where an expert defines mechanisms in the model using natural language which is interpreted and then assembled into an executable form, namely, the Ras Model and the MARM model. These models are particularly interesting as companions to the literature-derived models (e.g., the Ras Machine or the Neurofibromatosis model) in several ways, including the ability to diagnose issues with large-scale reading and assembly, and also relying on the literature-derived models to detect gaps in the expert-defined model based on limitations of its explanatory capabilities.
The COVID-19 model is currently the largest model in EMMAA - hardly surprising given the pace and quantity of publications on the topic - and is an example of a literature-derived model whose scope is defined fully based on the topic, not on specific entities or pathways. Meanwhile, the COVID-19 Disease Map model is an example of an expert-curated model that is imported into EMMAA, and then analyzed in EMMAA’s testing and query frameworks, demonstrating its capabilities to integrate community models.
Though somewhat of an outlier, the EMMAA Food Insecurity model serves to demonstrate the generalizability of EMMAA both conceptually and at a practical, working level. Namely, it uses a different literature source, different reading system, and a different assembly approach compared to the biology-oriented models to keep a model centered around scientific publications on food insecurity self-updating.
It is also important to discuss the robustness and accuracy of EMMAA models. While text mining and automated assembly of the scientific literature directly and in an automated fashion is inevitably imperfect, there are several strategies employed by INDRA/EMMAA to manage this. First, EMMAA models draw on multiple text mining systems as well as high-quality structured resources to aggregate evidence and corroborate statements from multiple sources. This is also the basis of establishing the belief score associated with each statement: the key metric for quantifying the underlying uncertainty for components of EMMAA models. We have now created a dedicated tab for browsing EMMAA models from the perspective of belief scores, and display belief scores for statements in all the relevant pages (see more details below). Ultimately, the transparency of these models, i.e., the ability of users to examine textual evidence and link back to original publications as provenance for every piece of knowledge integrated into models is crucial and sets this approach apart from classic expert-guided modeling.
In recent months, we have reported on sevaral approaches to strengthen EMMAA’s capabilities for dynamical modeling and simulation, including extended automated assembly pipelines, better integration with Kappa and BioNetGen, new dynamical property checking functionalities, and implementing support for the GroMET format (thereby connecting EMMAA to simulation frameworks developed for epidemiology models in ASKE-E). Going forward, we envision strengthening EMMAA further in this direction, working towards machine-assisted detailed analysis of models to gain insights about the underlying mechanisms.
Applying EMMAA model to COVID-19 therapeutics¶
The COVID-19 EMMAA model has continued monitoring and processing the literature being published - currently at a pace of around 315 papers per day - surrounding COVID-19. Since the beginning of 2021 the model has increased by 56% in terms of the number of papers processed. During this period, it also identified over 30,000 new unique causal statements, an increase of around 8%. Interestingly, while new relationships are still being frequently mentioned in the literature, the rate of new, unique, causal knowledge being published appears to be decreasing.
In terms of the model’s explanatory capability, the number of drug effects on coronaviruses it is able to explain (with the signed graph model) has grown from 1,814 to 2,132 this year, in other words, it has explained over 300 new drug effects based on new knowledge collected and assembled so far this year.
Review article on automated modeling¶
This month we submitted and revised a review article “From knowledge to models: automated modeling in systems and synthetic biology” which was accepted for publication, and is now available online at https://www.sciencedirect.com/science/article/pii/S2452310021000561.
The review introduces a conceptual framework for discussing levels of modeling automation with each level implying different roles for the human and the machine in the modeling process. The review discusses existing tools and approaches at each level of automation and provides. It also outlines the strengths and weaknesses of current modeling approaches at the different levels and discuss the prospect of fully automated fit-to-purpose modeling of biological systems.
Progress on inter-sentence causal connective extraction from text¶
We expanded our approach to generating a dataset of causally connected events in text. Our initial approach (described in last month’s report) involved finding sequential sentences in a paper that described paired directed interactions A->B and B->C and overwhelmingly produced positive examples, suggesting that sentence proximity alone could be used to deterministically identify causally transitive relationships. The lack of negative examples in the training data was a problem for development of a classifier.
During this period we expanded our search for relevant training examples (including negative examples) in three ways. First, we included undirected relationships (e.g., binding) in addition to directed relationships (e.g., activation or modification). Second, we considered multi-sentence relationships not only where the textually preceding event E1 causally preceded the textually subsequent event E2 (e.g., E1 causes E2) but also the reverse, where the causally downstream event was described first (E2 causes E1). Third, we searched for events up to three sentences apart.
This search produced a number of different types of examples not found previously. We manually curated 320 pairs of events and found 251 sentences where both events were correctly extracted; of these 148 were causally connected (positive examples) and 103 were not (negative examples). Pairs of events involving at least one undirected event were much more likely to be negative: for example, we found many cases where the binding of two proteins A and B was not causally connected to the activation of C by B, despite these events appearing closely in text. Interestingly, we also noticed examples of causal transitivity where a pair of events did not share a common node but were nevertheless causally connected. This can happen when an upstream event (e.g., the binding of EGF to EGFR) indirectly affects a downstream event (e.g., activation of ERK by MEK). The expanded dataset is being used by the University of Arizona team to develop a classifier to identify causal connections in text.
Integrating belief information in the EMMAA dashboard¶
We recently added a new tab on model dashboard to display belief statistics and browse statements based on their belief scores.
The following plot shows the distribution of belief scores in the COVID-19 EMMAA model. Having it visualized is useful for understanding the effect of using different belief scorers described in the previous report and of applying belief filters in the model assembly.
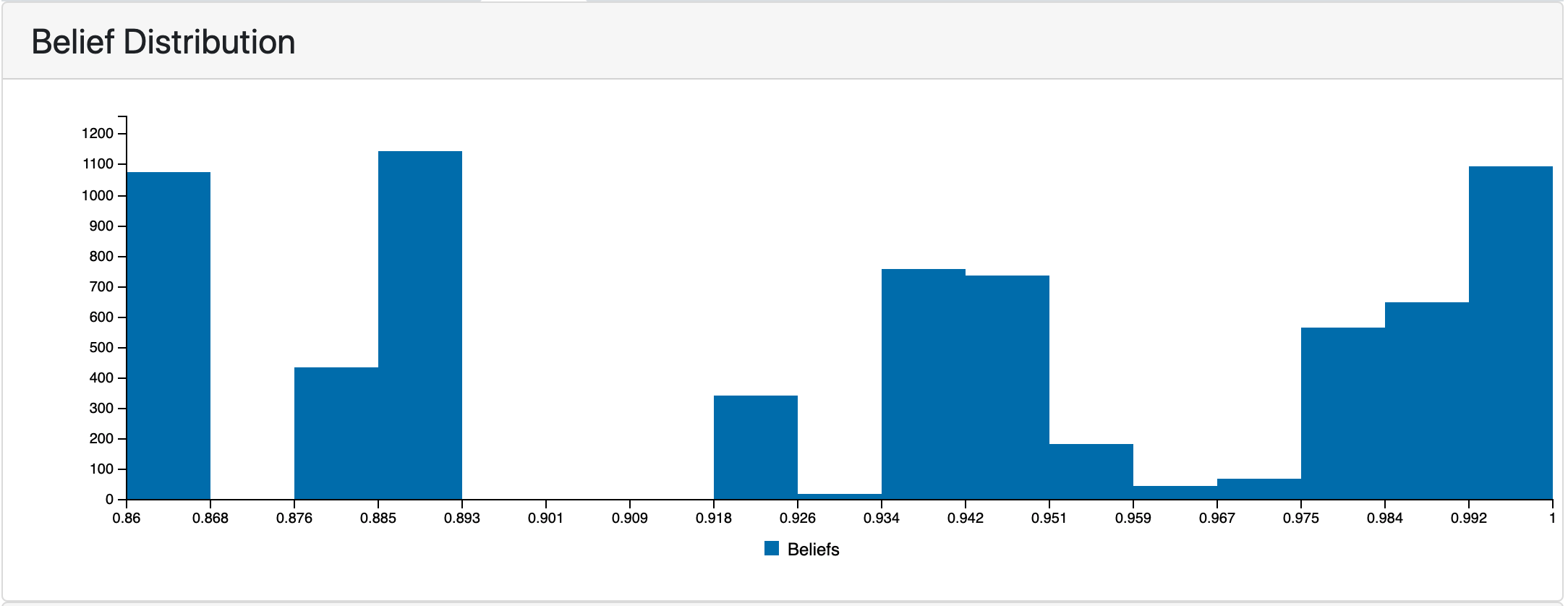
Belief scores distribution in RasMachine EMMAA model.
The next section in the belief tab shows the slider displaying the range of belief scores in a given model. A user can select a belief range and load the statements with the belief scores in that range. This gives a new way to prioritize the statements for the curation.
Belief scores range slider.
It is also possible to filter the statements to a given belief score range from the all statements page.
EMMAA model statements filtered to a given belief range.
Extending the ontology to epidemiology¶
To allow the Uncharted UI to use a single ontology covering both biology and epidemiology models, we aimed to extend the INDRA ontology with terms relevant for epi models. We found that the Infectious Disease Ontology (IDO) was an appropriate ontology to integrate since it contains terms such as “susceptible population” which correspond to commonly modeled nodes in epi models. However, in order to integrate IDO, we needed to implement a new module in INDRA to ingest ontologies in OWL format and expose their structure through an appropriate interface. Using this new OWL-ingestion module, we added nodes and relations from IDO to the INDRA Ontology graph and created a new export for use in the Uncharted UI.
STonKGs¶
The transformers and attention architectures have reinvigorated large scale language models through BERT and its derivatives. We are part of a collaboration with the Fraunhofer Institute to develop a Sophisticated Transformer trained on biomedical text and Kknowledge Graphs (STonKGs), a joint knowledge graph and language model that relies on the following cross-modal attention mechanism:
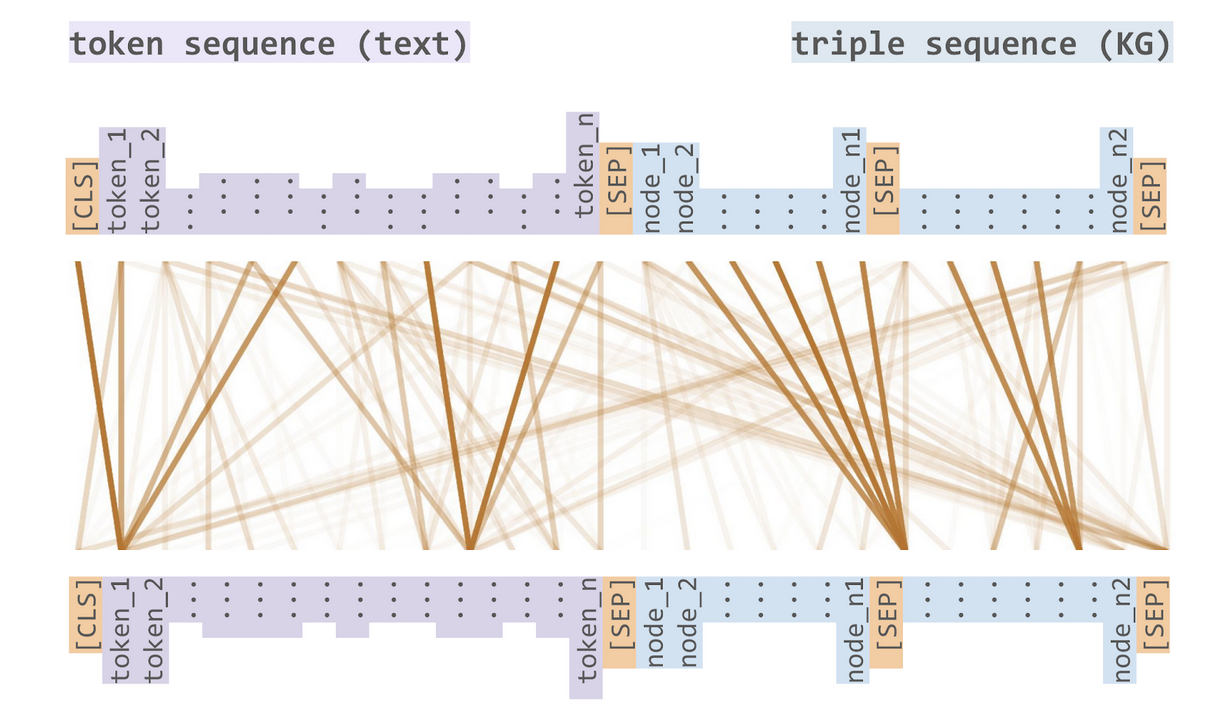
The cross-modal attention mechanism in the STonKGs model enables joint learning over INDRA statements and their associated textual evidences
This model is able to take an INDRA statement and its associated text then generate a dense Euclidean vector representation that can be used for downstream machine learning tasks. We have prepared a dump of the INDRA database in order to pre-train this model and suggested several downstream “fine-tuning” binary/multi-class classification tasks on which STonKGs could be evaluated:
| Task | Description | Example |
|---|---|---|
| Polarity | Directionality effect of the source node on the target node | “HSP70 […] increases ENPP1 transcript and protein levels” (PMID:19083193) |
| Interaction Type | Whether it is known to be a physical interaction between the source and target node | “SHP repressed […] transcription of PEPCK through direct interaction with C/EBPalpha protein” (PMID:17094771) |
| Cell Line | Cell line in which the given relation has been described | “We show that upon stimulation of HeLa cells by CXCL12, CXCR4 becomes tyrosine phosphorylated” (PMID:15819887) |
| Disease | Disease context in which the particular relation occurs | “ […] nicotine […] activates the MAPK signaling pathway in lung cancer” (PMID:14729617) |
| Location | Cellular location in which the particular relation occurs | “The activated MSK1 translocates to the nucleus and activates CREB […].” (PMID:9687510) |
| Species | Species in which the particular relation has been described | “Mutation of putative GRK phosphorylation sites in the cannabinoid receptor 1 (CB1R) confers resistance to cannabinoid tolerance and hypersensitivity to cannabinoids in mice” (PMID:24719095) |
Ultimately, the model showed ability to learn and predict within the training/testing split across all tasks better than only using purely network-based prediction methods or purely text-based prediction methods.
Further investigation is necessary to assess its overfitting to the underlying text mining systems, such as REACH, by generating additional curated corpora for each task that had not already been read by REACH.
PyKEEN Updates¶
Improvements to Link Prediction Evaluation Metrics¶
The common evaluation metrics used in the link prediction task for knowledge graph embeddings (e.g., mean rank (MR), mean reciprocal rank (MRR), and hits at k) are not comparable for knowledge graphs of varying number of entities, relations, and triples. This poses a problem a as we move to apply knowledge graph embedding models to biomedical knowledge graphs because we are interested in comparing different formulations (e.g., using just knowledge from databases vs. INDRA’s entire knowledge graph).
Berrendorf et al. (2020) proposed the adjusted mean rank, which normalized the value based on the expected value. We have derived closed form expectations for the mean reciprocal rank and hits at k and implemented their corresponding adjustments in PyKEEN.
Further we developed an alternative metric to the hits at k that uses a smooth logistic sigmoid instead of a discrete step function in order to mitigate some of its biases, including its applicability to graphs of varying sizes.
Improvements to Loss Functions¶
The binary cross entropy loss, softplus loss, margin ranking loss, and non-self adversarial negative sampling loss have proven to be the most popular in knowledge graph embedding models. However, there are deep theoretical relationships between them, such as the alleged equivalence between the softplus loss and binary cross entropy loss with sigmoids, that have been relatively unexplored. We improved the programmatic design to generalize and identify some of these concepts, as well as provide implementations of the double margin loss and focal loss which we believe might be more valuable for applications to biological networks.
The double loss is given as:
The focal loss is given as
with \(p_t = y \cdot p + (1 - y) \cdot (1 - p)\), where \(p\) refers to the predicted probability, and y to the ground truth label in \({0, 1}\).
Non-Parametric Baseline Models¶
Many supervised machine learning methods use y-scrambling or similar methods for generating null models against which the true model can be compared. Because knowledge graph embedding models are so time-consuming to train, comparison to a null model is often omitted in both theoretical and practical work. We have developed two non-parametric baseline models based solely on entity and relation co-occurrence that require no training.
For the marginal distribution model, to predict scores for the tails, we make the following simplification of \(P(t | h, r)\):
Surprisingly these perform very well, and ultimately provide a minimum threshold that any more knowledge graph embedding model must surpass. The results are available here.
BioCreative participation and new Walkthrough Tutorial¶
We continued working on the BioCreative challenge on interactive COVID-19 text mining tools. Our proposal “A self-updating causal model of COVID-19 mechanisms built from the scientific literature” was accepted for participation. This month, we prepared a system description document, recruited test users, and created a new tutorial for using EMMAA’s COVID-19 model.
This “Walkthrough tutorial” illustrates EMMAA’s key features through the COVID-19 model using a series of short videos, descriptions, and to-do actions. Each section also describes relevant background. The tutorial can be found here: https://emmaa.readthedocs.io/en/latest/tutorial/index.html.