ASKE Month 5 Milestone Report: Lessons Learned¶
Here we summarize some of the high-level lessons we learned about large-scale machine-assisted model assembly and analysis over the course of developing EMMAA. Overall, we strongly believe that through an attempt to automate scientific modeling, we can gain substantial insight into the way human experts work with models of complex systems.
Automated model assembly: the challenge of defining scope and context¶
The initial development of EMMAA focused on deploying an automated model assembly pipeline to generate models specific to the various cancer types catalogued in the cancer genome atlas (TCGA). Collectively these models would constitute an “Ecosystem” of self-updating, context-specific models that could be used to answer mechanistic queries relevant to specific diseases. Context specificity was necessary because the answer to queries (e.g. “What is the effect of EGFR inhibition on cell growth?”) differ depending on the specific gene expression pattern and mutation profile of a particular cancer type.
Our initial approach to enforce the context-specificity of automatically assembled models is described here and is centered on the genetics of specific cancer types. Frequently mutated genes in specific cancers were used as search terms to query Pubmed for publications which were then processed with machine reading tools and assembled into models along with information from curated databases.
Subsequent model testing highlighted a key shortcoming of this approach: tests of well-known biochemical pathways would fail in nearly all models because the limitations imposed on model scope (in the interest of context specificity) resulted in many key genes being omitted.
In an effort to expand models to incorporate key “backbone” genes while still retaining context specifity we then implemented two alternative approaches.
Run heat diffusion over our biological knowledge network to identify genes that were highly connected to the cancer-specific genes;
Query Reactome, a high quality database of biological pathways, for pathways containing the disease genes, and incorporate all genes from these pathways into the model.
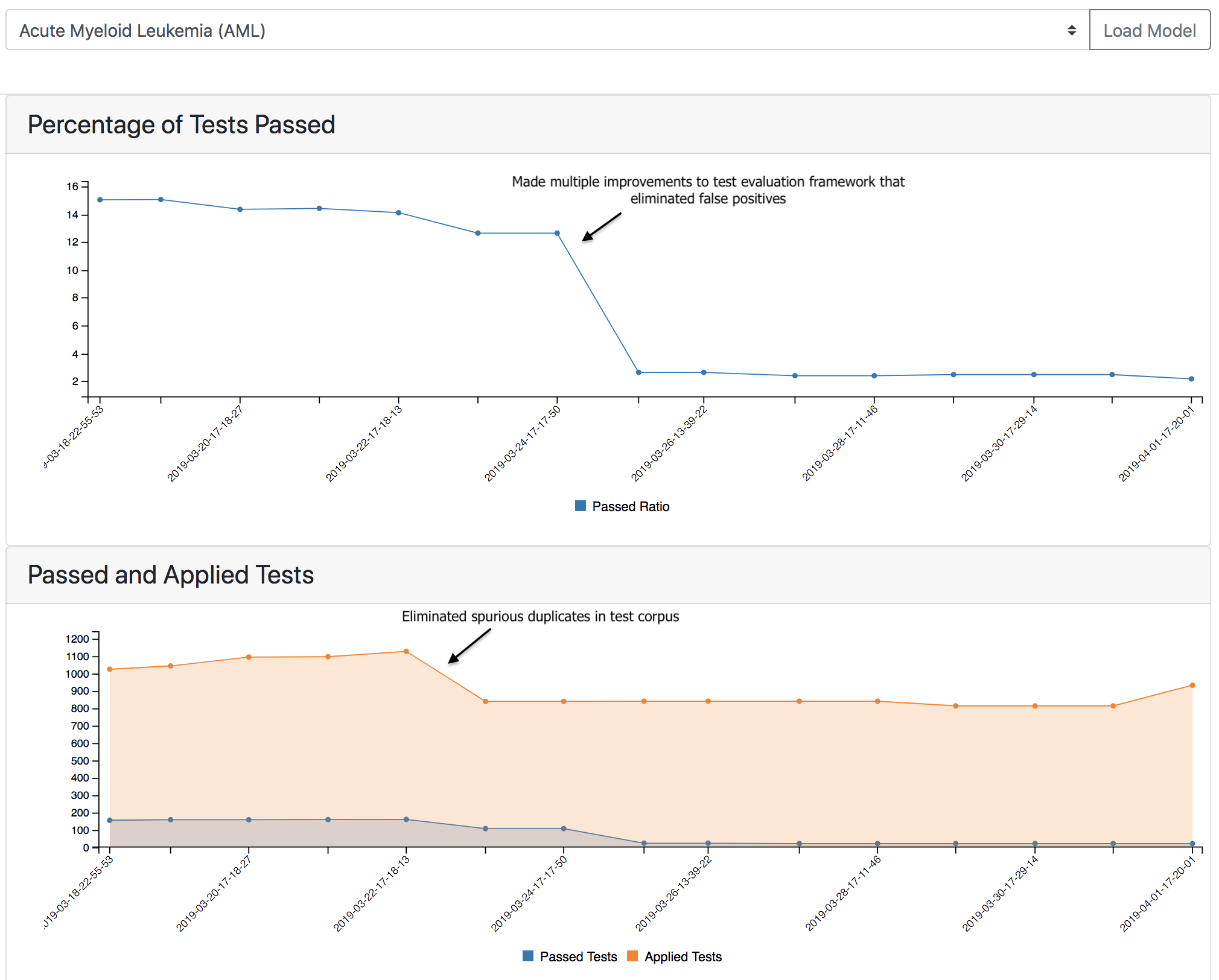
We found that the latter approach involving Reactome was more effective at eliminating mechanistic gaps than heat diffusion, which tended to highlight irrelevant genes based peculiarities of the knowledge network structure. However, even with the automated Reactome-based approach we found that models had a very low ratio of passing tests, and glaring mechanistic gaps: for example, the melanoma model passed only 4% of tests from the BEL Large Corpus, and omitted MAP2K1, a protein immediately downstream of (the frequently mutated gene) BRAF and a validated target in melanoma.
We therefore explored an alternative approach, in which models would be made specific to biochemical pathways rather than cancer types, a la the original Ras Machine. We found that the first iteration of this model had a much higher pass ratio of 34%, suggesting that models built and limited in scope in this way were more likely to have the internal integrity required for answering mechanistic queries.
Despite this improvement, the central problem of capturing model context remains: even if an automatically assembled model contains the genes relevant to a specific disease does not imply that it can answer a mechanistic query in a context specific way. For example, the Ras pathway is involved in many cancer types, not least in lung cancer and melanoma, yet the effects of intervening in the pathways differ between the two diseases. A key remaining challenge is to develop a system that can pull in the relevant data (e.g., gene expression, mutations) to contextualize structurally identical models, and make use of this data during analysis to reach context-specific conclusions.
Automated model analysis: benefits of automated model validation¶
With respect to model analysis, the first key lesson learned is how valuable the process of automated testing is for developing model assembly systems such as INDRA and EMMAA. By coupling large-scale automated reading and assembly with automated testing and analysis, the strengths and weaknesses of the reading/assembly machinery itself are clearly exposed. Over the course of monitoring daily updates to the disease models and browsing test results, we were able to identify bugs and other opportunities for improvement in a highly efficient and targeted way. The image below illustrates the effect of some of these improvements as they affected the number of applied and passed tests:
Test-driven modeling¶
A key observation that we have made during the development of EMMAA is of the value of automated model testing not only as a means of post-hoc model validation, but also to support test-driven modeling. That is, the construction (in part manual) of scientific models based on a corpus of qualitative experimental constraints. This is by analogy with test-driven development in software engineering in which the tests are written first, and program features are only added to satisfy the tests.
During this reporting period, we have explored test-driven modeling by manually building a model of a core subset of the Ras signaling pathway. The model is built using natural language via INDRA and TRIPS as described here; the automated assembly of the natural language sentences yields a model with semantic annotations enabling subsequent testing and analysis. The model is exposed in the EMMAA dashboard as the “Ras Model”. The initial model consisted of a set of roughly 60 natural language sentences and was roughly doubled in size through an iterative process of expansion and refinement that was driven by model testing.
We have found that the test-driven modeling approach has a number of advantages for the construction of scientific models. First, the approach to scientific modeling in many fields is to use a formal model to encode a specific hypothesis about a particular phenomenon. These “fit-to-purpose” models are useful tools for answering specific scientific questions but they are rarely reusable and are biased toward a particular explanation. With test-driven modeling, the growth of the model is empirically driven by the observations that match the scope of the model, independent of any specific problem. In extending the Ras model to satisfy tests from the BEL Large Corpus, we repeatedly found it necessary to add in underappreciated or noncanonical mechanisms. For example, it is well known that EGFR activation leads to the phosphorylation and activation of SRC; but it is also the case that SRC phosphorylates and potentiates the activation of EGFR. Similarly, AKT1 both phosphorylates and is phosphorylated by MTOR. In typical practice, a modeler would not incorporate all of these influences unless it was their specific intention to investigate crosstalk, feedback, or other aspects of the overall mechanism that deviate from a simple linear pathway. The process of test-driven modeling brought to the forefront how common these processes are.
Second, just as in software development, test-driven modeling helps the modeler avoid decorating a model with details that are not essential to improving overall performance. This helps to avoid modeling quagmires in which a modeler attempts to encode everything known about a process in maximum detail. The existence of a set of tests, and the iterative development process that EMMAA enables (serving here as a tool for continuous integration of models), dramatically improves the efficiency of building high quality, reusable models.
Third, test-driven modeling helps build insight into how a model works, as well as highlighting serendipitous and potentially unexpected implications of particular mechanisms. During the test-driven development of the Ras Model, there were several instances where adding a small extension to the model to address an issue that appeared to be local to the two proteins resulted in several additional tests passing, that involved long-range causal influences. For example, fixing a reaction involving MTOR and PPP2CA resulted in three tests passing, each highlighting the negative feedback from MTOR back to upstream IGF1R signaling via IRS-1.
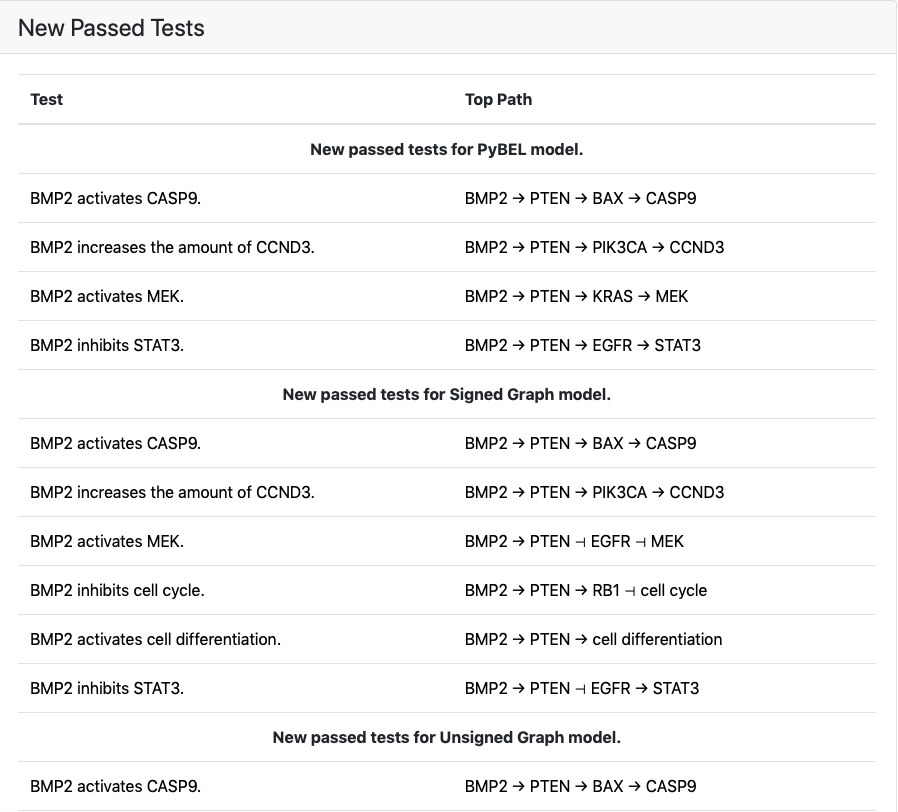
The screenshot of the EMMAA dashboard test results page for the curated Ras Model, shown below, highlights the iterative process of test-driven model refinement and expansion.
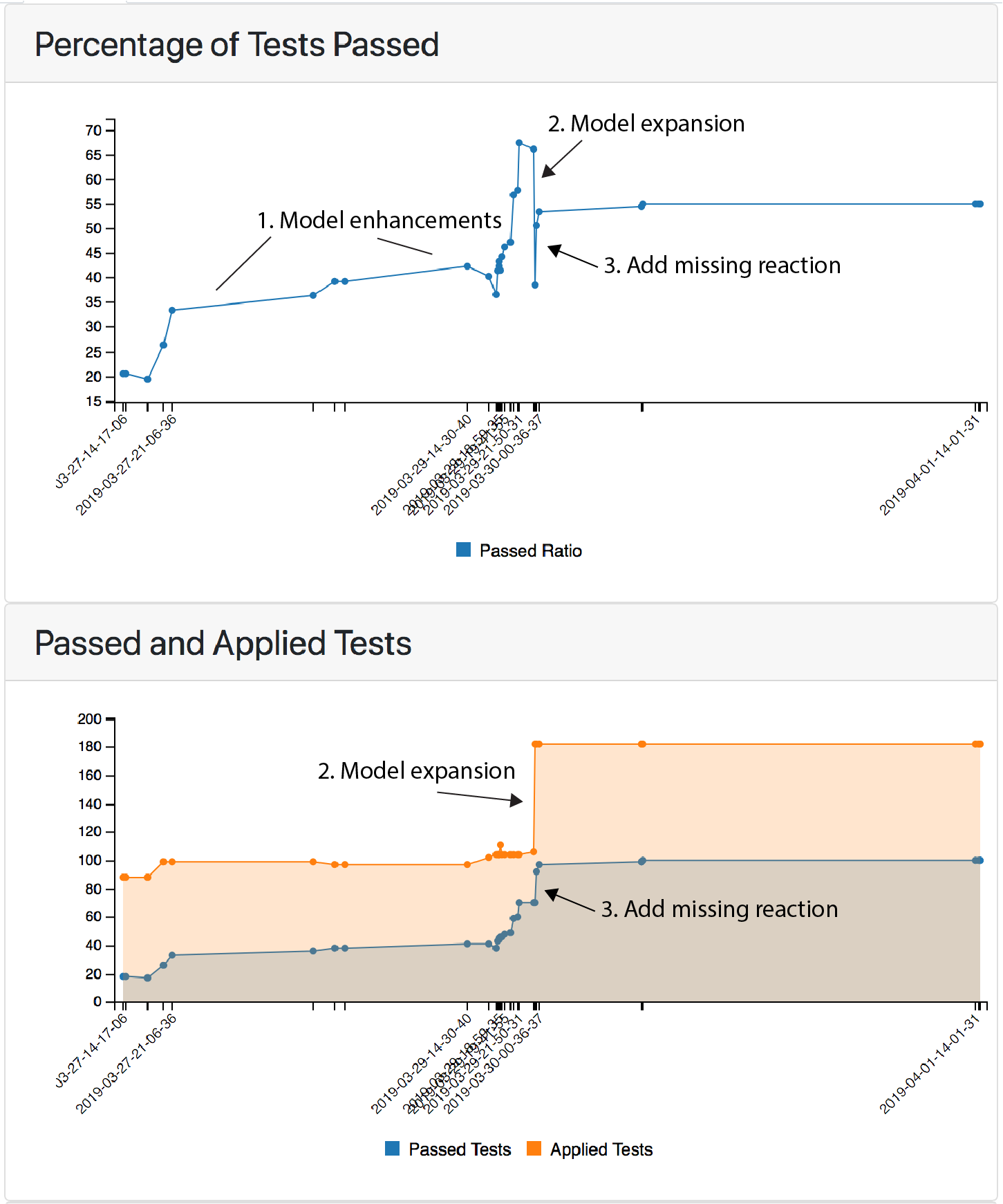
The bottom plot shows the total number of applied tests over time, along with the number of passing tests; the top plot tracks changes in the percentage of passing tests. The initial process of model refinement is shown by (1), in which the initial model was subject to testing and then progressively refined over time. During this process the pass ratio grew from roughly 20% to 67%. At this point, the model was expanded to include the well studied signaling proteins EGF and EGFR. This nearly doubled the number of applied tests (2, bottom plot), but since relatively few of these new tests passed, the pass ratio dropped to ~35%. Importantly, these new tests were applied automatically by EMMAA as a consequence of the expansion in model scope. Inspection of the model highlighted the fact that EGFR was disconnected from many of its downstream effectors; addition of only a single statement (connecting EGFR to SOS1, which was already in the model for its role downstream of IGF1R) led to a large number of the new tests passing, boosting the pass ratio back to over 50% (3, both plots).
Exploiting the bidirectional relationship between models and tests¶
During the development of EMMAA we have come to appreciate the benefits of treating the information flow between models and tests as symmetric and bidirectional.
For example, manually curated tests can be used to validate automatically assembled models, or the other way around: curated models validating automatically extracted observations. In our initial work, we focused on the application of curated experimental observations (from the BEL large corpus) to automatically assembled mechanistic models. We described above how applying these tests to the Ras Machine model helped us to identify issues in our automatic model assembly pipeline that had been latent for years. We now also see the value in automatically collecting tests and using high-quality curated models to evaluate the plausibility of the test observations themselves. For example, in the development of the Ras Model, we found that a surprising proportion (over 15%) of the tests in the BEL Large Corpus were incorrectly curated. These test errors were inadvertently highlighted when the model failed to pass them. We imagine that observations derived from a noisy source (such as machine reading) could be subjected to checking by one or more high-quality models, with the model establishing the likelihood that a finding resulted from a machine reading error. It is also possible to imagine that in fields where models are mature, new scientific findings could be automatically subjected to model-driven evaluation, highlighting the ways in which they either support or contradict established models.